SCIENTIFIC INTENT PARSER
"Find genes causing drug resistance in my cohort"
▼
KNOWLEDGE-DRIVEN METHOD SELECTOR
"Use DESeq2 for low counts, Cox for survival data"
BioBuild's Moat
▼
PIPELINE SYNTHESIZER
Step 1
→
Step 2
→
Step 3
→
Step 4
▼
▼
▼
▼
EXECUTION ENGINE
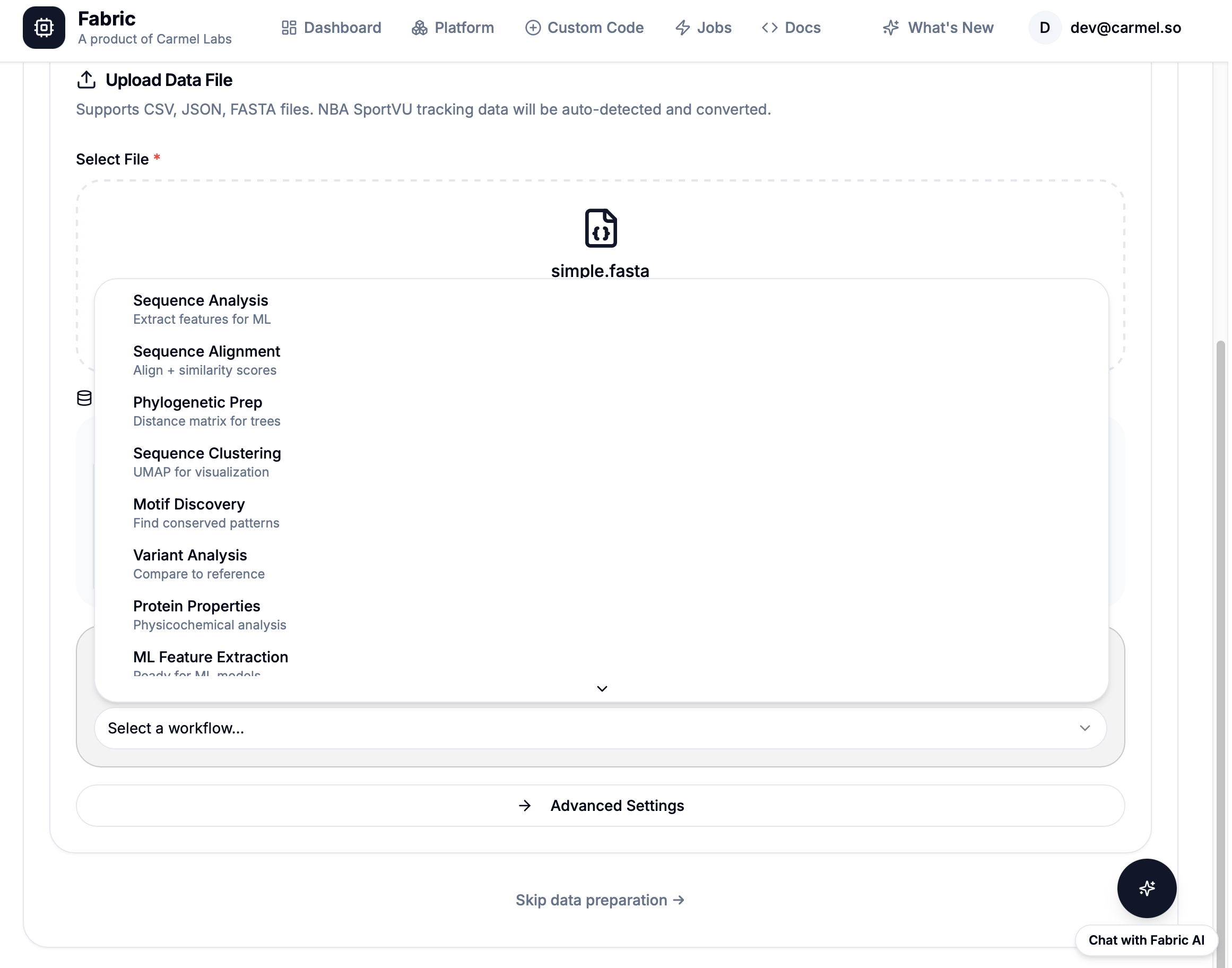
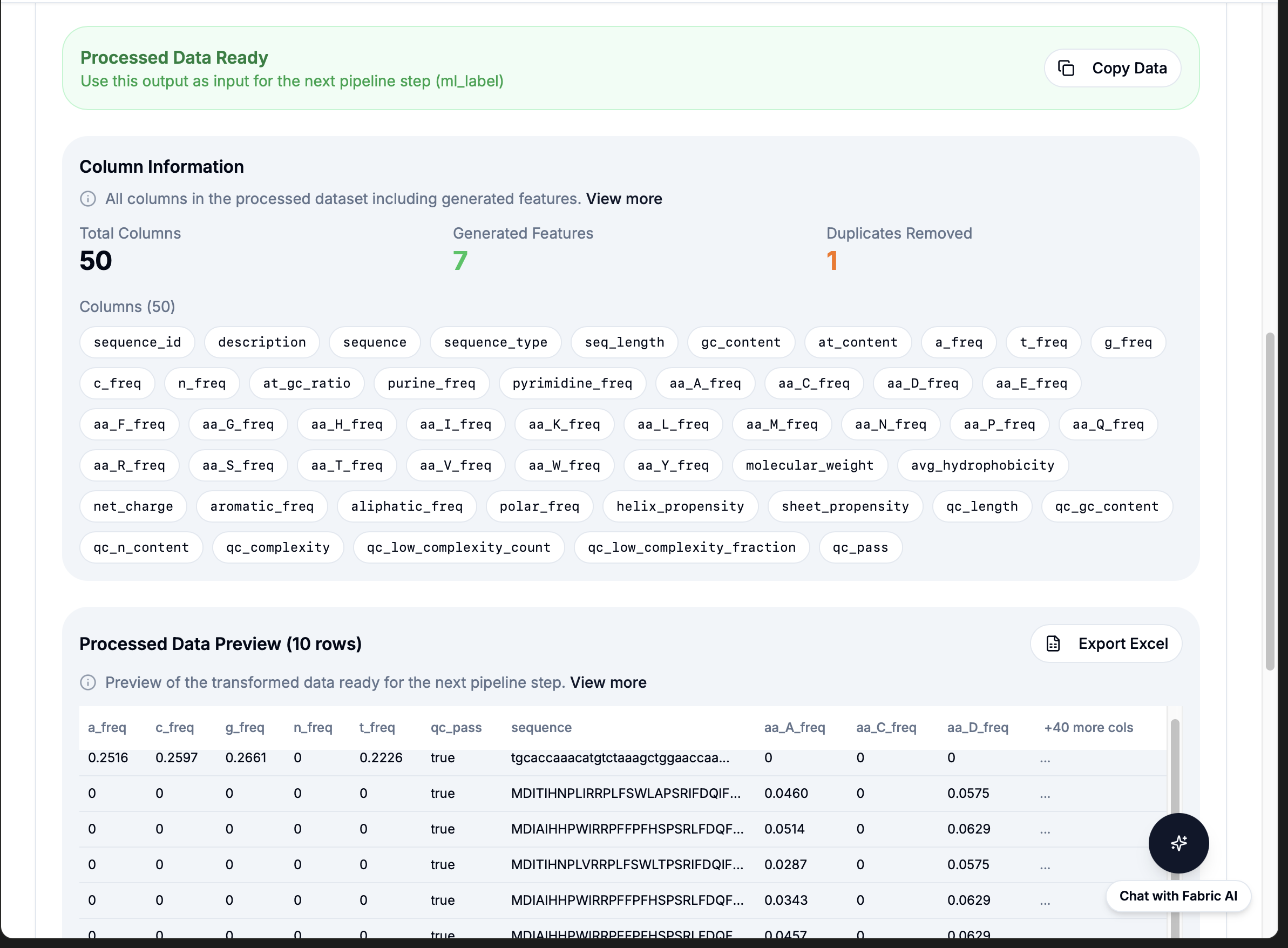
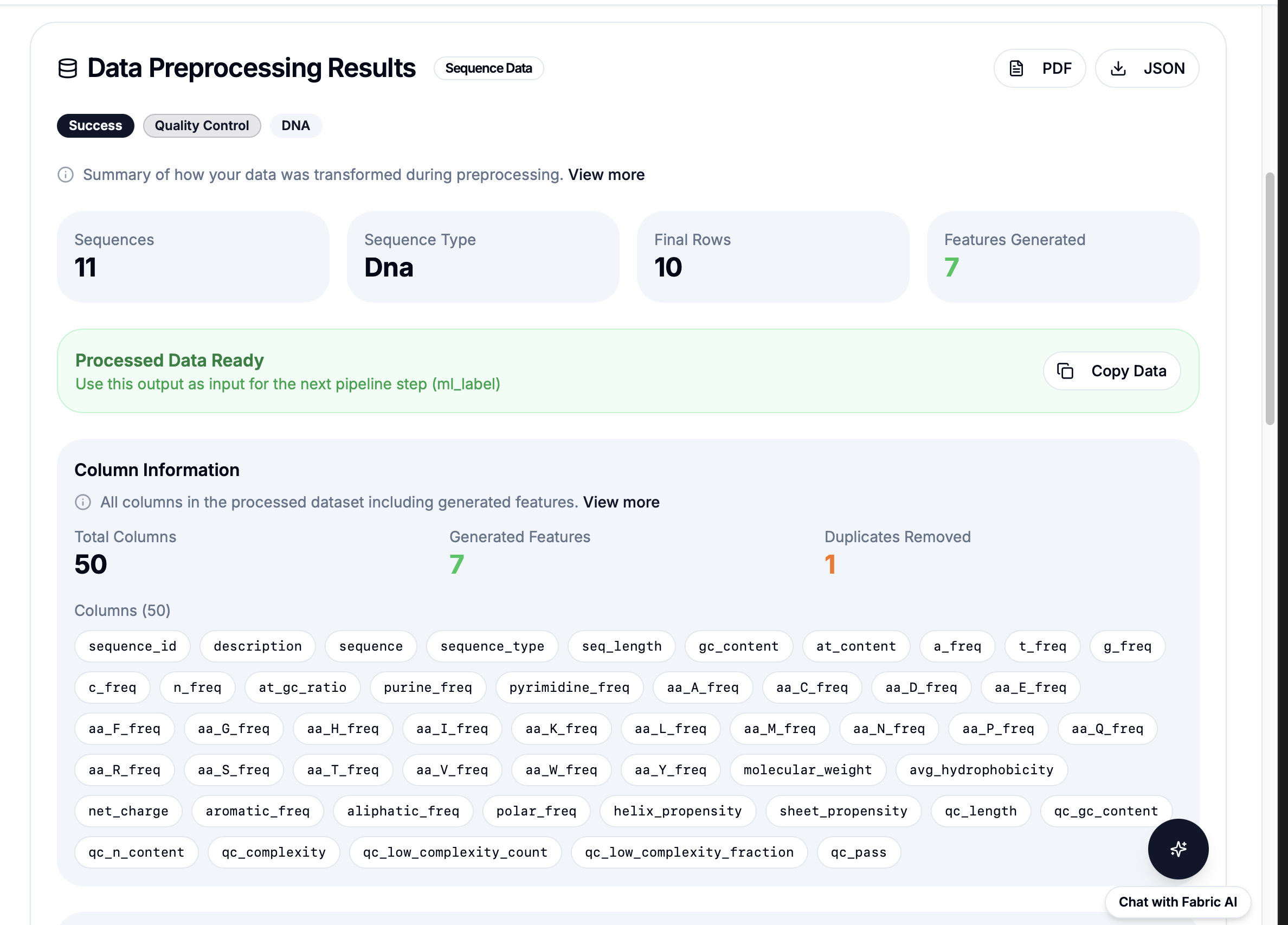
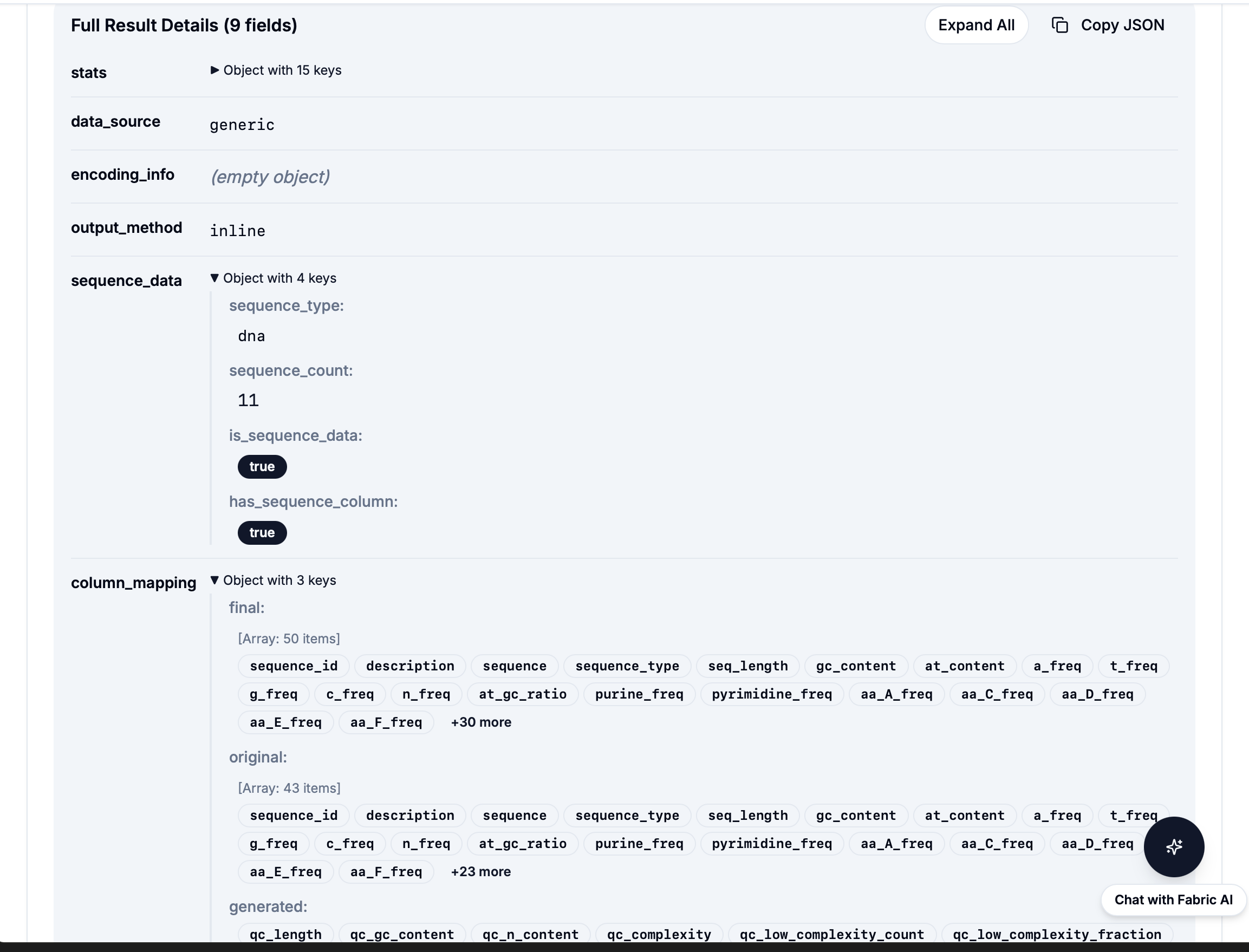
powered by Fabric: 21 Pre-built Bioinformatics Workflows
Sequence
GC, Align, Translate
Gene Analysis
Codon, ORF, Tm
Protein
Domains, Structure
Quality
Filter, QC, Stats
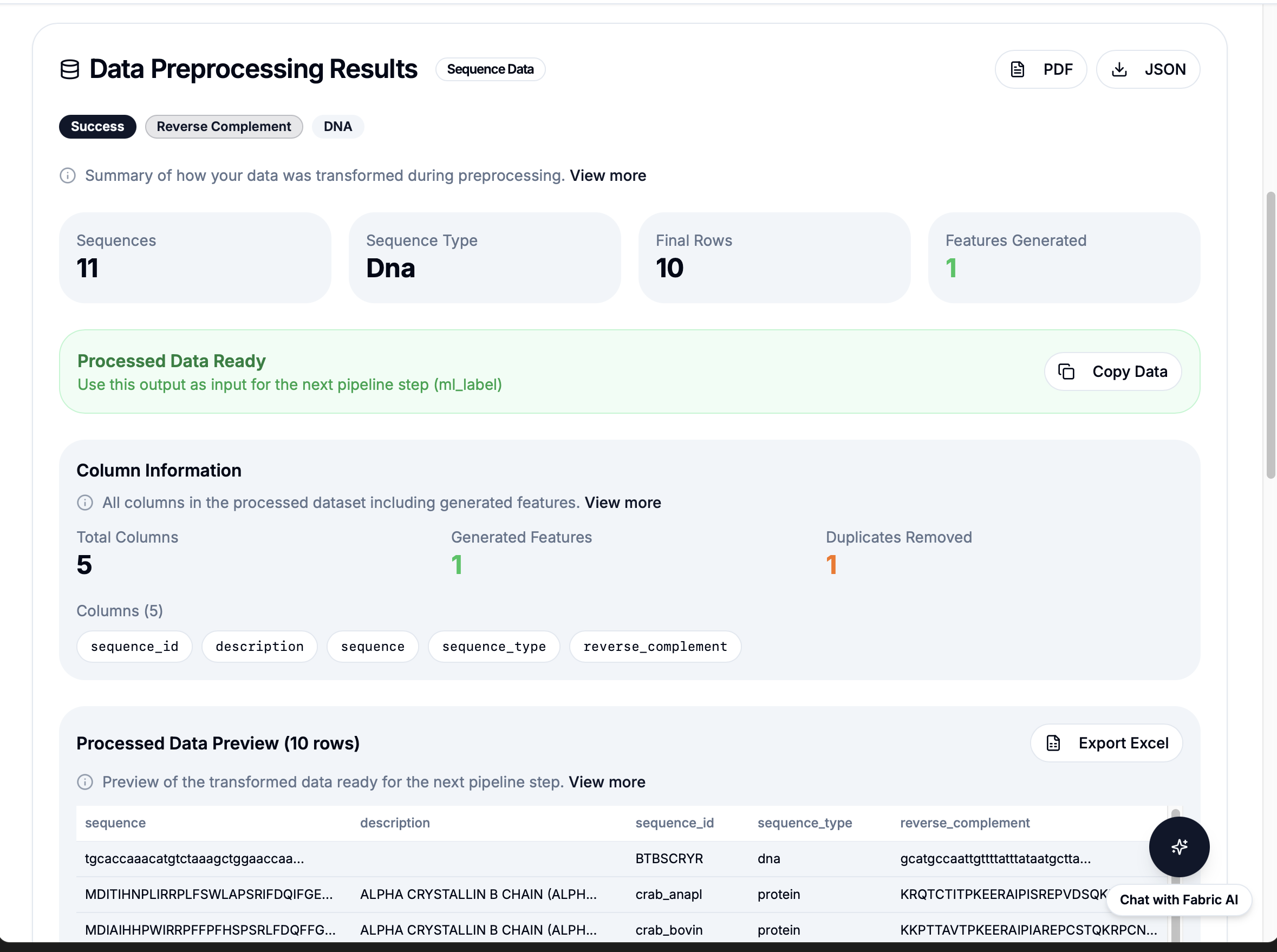
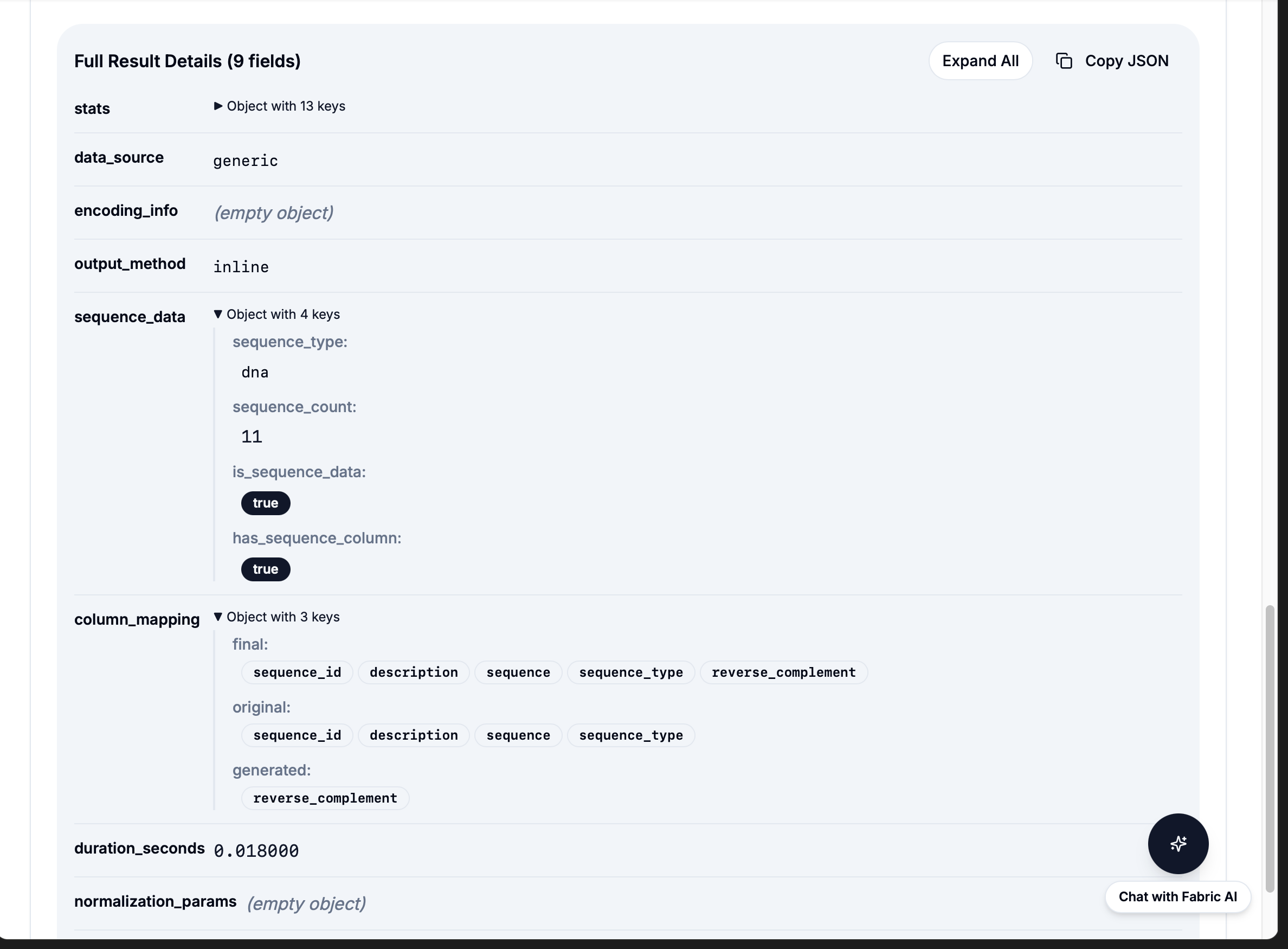
Simple API: workflow: "reverse_complement" then results in under 1 second
[ Distributed Agent Network / Auto-scaling / 100% Reliable ]
▼
▼
▼
▼
RESULTS + PROVENANCE + VISUALIZATION
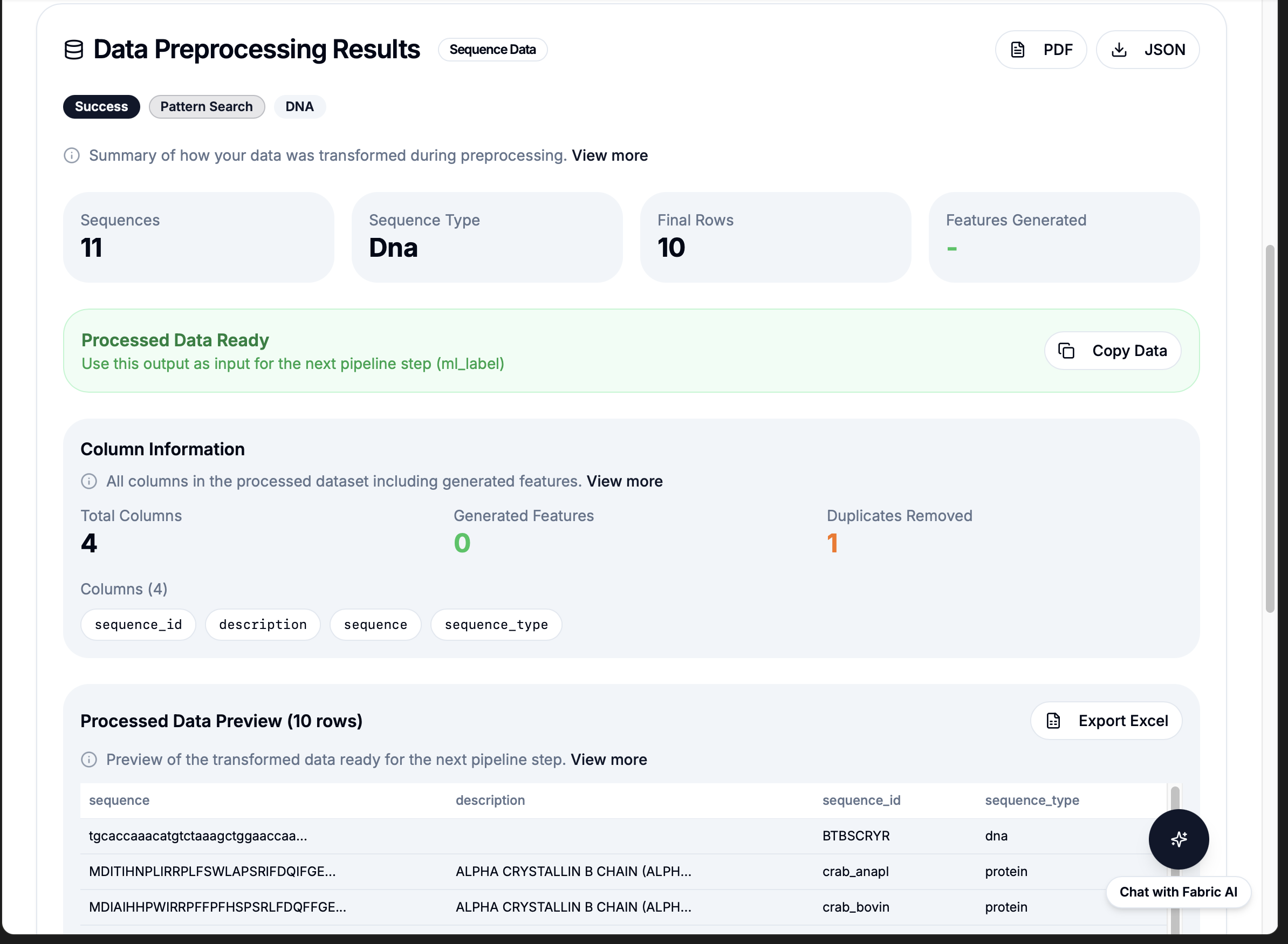
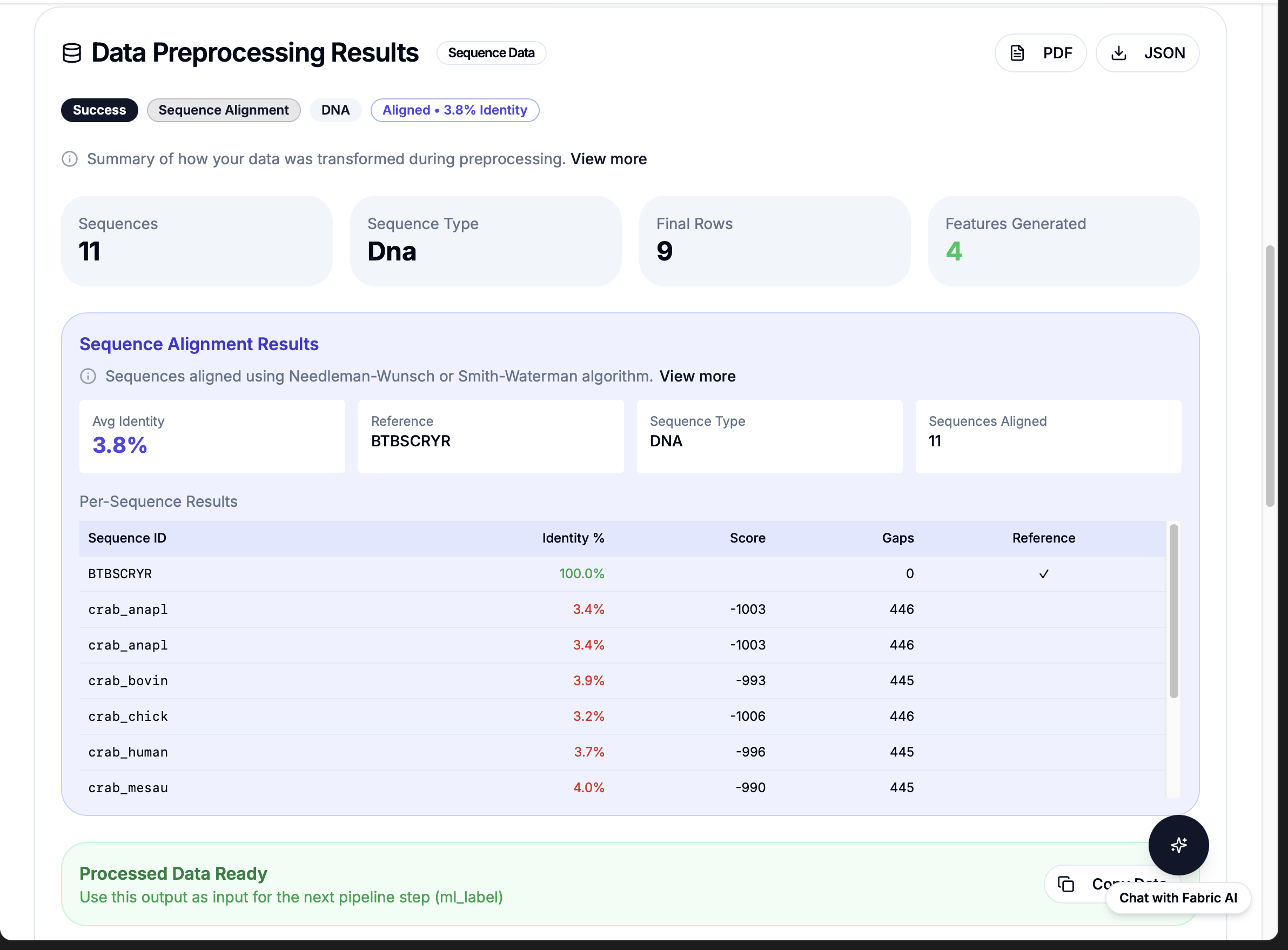
Proof of Concept: Live Demo Results
21
Workflows
Pre-built bioinformatics operations ready to call
<1s
Execution Time
Per workflow call
100%
Reliability
No syntax errors, no retries, production tested
Integration model: BioBuild's AI understands "calculate reverse complement" then calls
workflow: "reverse_complement" then Fabric executes instantly then results returned. No code generation needed.